
Sterilization by Liquid Filtration: How It Works and When to Use It
Sterilization by liquid filtration removes microorganisms from a heat-sensitive solution by passing it through a membrane filter with a pore size small enough to retain bacteria — most commonly 0.22 µm. Unlike steam, EtO, gamma, or hydrogen peroxide, filtration does not kill microorganisms; it physically separates them from the fluid. It is the standard sterilization method for vaccines, parenterals, ophthalmic products, cell culture media, and any other heat-sensitive liquid that cannot be autoclaved.
This article explains how membrane filtration achieves sterility, what it can and cannot remove, integrity testing, and how filtration compares to autoclave sterilization for liquid loads.
What Is Filtration Sterilization
Filtration sterilization is a mechanical separation process. A liquid is forced (by positive pressure or vacuum) through a porous medium that retains particles larger than its rated pore size. Microorganisms larger than the pore — bacteria and most fungi — are physically retained on or in the membrane. The filtrate, free of viable organisms, exits as a sterile product if the entire filter assembly is integral.
Two key consequences follow from this mechanism:
- Filtration does not kill microorganisms. They are retained alive in the filter and must be inactivated separately (e.g., by autoclaving the spent filter).
- Filtration retains by size, not by killing chemistry. Anything smaller than the pore — most viruses, mycoplasma, prions, endotoxins — passes through unless a finer filter or different mechanism is used.
How Membrane Filtration Works
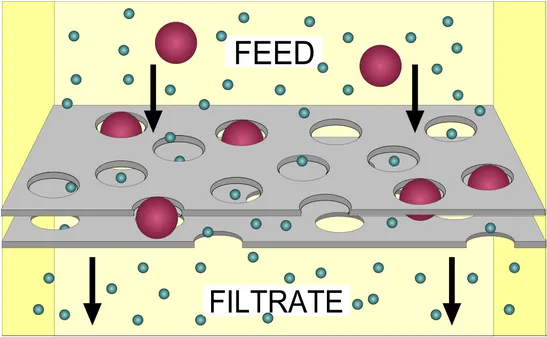
Modern sterilizing-grade filters are membrane filters with tightly controlled pore distribution. The defining standard is 0.22 µm (sometimes 0.2 µm — the same membranes are sold under both labels), which retains the standard challenge organism Brevundimonas diminuta at >10⁷ CFU/cm² of filter area, the regulatory benchmark for "sterilizing-grade" performance.
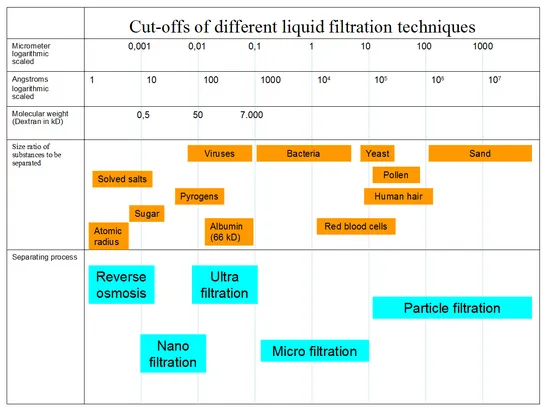
| Pore size | Retention | Use case |
|---|---|---|
| 0.45 µm | Most bacteria, but some pass | Bioburden reduction; not sterilizing |
| 0.22 / 0.2 µm | Sterilizing grade — retains B. diminuta | Standard for sterile filtration of pharmaceuticals, media |
| 0.1 µm | Mycoplasma | Cell culture media, mycoplasma-free filtration |
| 0.04 µm and smaller (UF/NF) | Some viruses, large proteins | Virus reduction, ultrafiltration |
Membranes are made from polymeric materials selected for chemical compatibility:
- PES (polyethersulfone) — broad chemical compatibility, low protein binding, high flow
- PVDF (polyvinylidene fluoride) — hydrophilic or hydrophobic, broadly compatible
- Cellulose acetate / nitrate — older membranes, lower protein binding, narrower compatibility
- Nylon — low cost, narrower compatibility
- PTFE — hydrophobic, used for venting and gas filtration, requires pre-wetting for liquid use
Filters can be flat-sheet (in syringe filters or capsules) or pleated cartridges for higher-throughput in-line sterile filtration.
Types of Filters Used
| Filter type | Construction | Application |
|---|---|---|
| Syringe filter | 13–25 mm disc in disposable housing | Small-volume sterilization at the point of use (a few mL to ~100 mL) |
| Capsule filter | Self-contained polymeric capsule | Mid-volume sterile filtration; single-use bioprocessing |
| Cartridge filter | Pleated membrane in stainless or plastic housing | High-volume in-line sterilization in pharmaceutical manufacturing |
| Bottle-top vacuum filter | Disc membrane on receiver bottle | Sterilization of media, buffers in the lab |
Older filter types — Seitz pad filters, sintered glass filters, and unglazed-ceramic candle filters — were once used for bacterial filtration but have been largely replaced by polymeric membrane filters with defined and reproducible pore size.
What Can Be Sterilized by Filtration
Filtration is the standard sterilization method for heat-sensitive aqueous solutions:
- Pharmaceuticals: parenteral injectables, ophthalmic solutions, biologics
- Vaccines and serum-containing products
- Cell culture media and supplements
- Antibiotic and protein solutions
- Buffers and reagents for laboratory use
- Some consumer products: certain cosmetic preparations, beverages
It is not appropriate for:
- Suspensions or particulate-laden fluids (clog the filter)
- Viscous liquids beyond the filter's pressure rating
- Solutions where the sterilizing membrane adsorbs the active ingredient (preservative or low-concentration drug binding)
- Viral inactivation at 0.22 µm (most viruses pass through)
Filtration vs Autoclave Sterilization for Liquids
Both methods sterilize liquids, but they are appropriate for different load types.
| Factor | Liquid filtration (0.22 µm) | Autoclave (steam) |
|---|---|---|
| Mechanism | Mechanical separation | Moist heat protein coagulation |
| Temperature on the load | Ambient (or controlled) | 121 °C |
| Heat-sensitive products | Yes — preserves activity | No — denatures proteins, alters pH |
| Viral removal | Generally no | Yes (heat inactivation) |
| Mycoplasma removal | Requires 0.1 µm | Yes |
| Particulate-laden fluids | No (clogs) | Yes |
| Validation | Integrity test + bacterial challenge | Cycle parameters + biological indicator |
| Scale | Lab to large pharma | Lab to large industrial |
| Cost per litre | Filter cost-driven; higher per litre | Lower for large volumes |
| Speed | Continuous flow | Batch cycle (~30–90 min) |
For heat-stable solutions (saline, simple buffers, glassware-rinse water), autoclave (steam) sterilization is faster, cheaper, and more thorough — and inactivates viruses. For heat-sensitive solutions where activity must be preserved (proteins, vaccines, antibiotic stocks, media containing serum), filtration is the only viable option.
Validation and Quality Control
Sterile filtration is validated through three pillars:
1. Bacterial Retention Validation
A 0.22 µm filter is challenged with Brevundimonas diminuta (ATCC 19146) at ≥10⁷ CFU/cm² of effective filter area, with the actual product or a representative diluent. Sterilizing-grade is demonstrated when no organisms pass through under the filter's specified flow and pressure conditions.
2. Filter Integrity Testing
Each filter assembly must be tested before and/or after use to confirm that it has not been damaged or installed improperly. Two non-destructive methods:
- Bubble point test — measures the minimum gas pressure required to displace liquid from the largest pore. A reading at or above the manufacturer's specification confirms integrity.
- Diffusion (forward flow) test — measures gas diffusion through a wetted filter at a defined pressure. Reading must be below the manufacturer's specification.
3. Process Validation
Full process validation under PDA Technical Report 26 and FDA / EMA guidance covers fluid–filter compatibility (extractables, leachables, adsorption), sterilization of the filter assembly (typically by autoclave or in-line steam), aseptic connection, and downstream sterility.
Limitations
- Most viruses pass through 0.22 µm. Virus reduction requires ultrafiltration, dedicated virus-removal filters (e.g., 20 nm), or other methods (heat, chemical, irradiation).
- Mycoplasma can pass 0.22 µm. A 0.1 µm filter is required for mycoplasma-free filtration of cell culture media.
- Endotoxin (LPS) passes through. Endotoxin removal requires specific affinity media (charged depth filters, polymyxin columns, ultrafiltration).
- Filter adsorption can deplete low-concentration active ingredients (preservatives, low-dose drugs); fluid–filter compatibility must be tested.
- Particulates can blind the filter; pre-filtration with coarser membranes may be needed.
- No retained kill record — unlike a heat cycle, filtration leaves no time-temperature record; integrity of the assembly is the proof.
Regulatory Standards
- PDA Technical Report 26 — Sterilizing filtration of liquids (the industry's principal validation reference).
- USP <1211> — Sterility assurance of compendial articles, including sterile filtration.
- USP <797> — Pharmaceutical compounding; sterile preparations.
- EU GMP Annex 1 — Manufacture of sterile medicinal products. Specifies sterile filtration requirements for non-terminal sterilization.
- FDA Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing — Current Good Manufacturing Practice (2004).
- ISO 13408 series — Aseptic processing of health care products, with specific parts on filtration.
FAQ
What pore size is used for sterilization filtration?
The standard sterilizing-grade pore size is 0.22 µm (sometimes labeled 0.2 µm), validated against Brevundimonas diminuta at 10⁷ CFU/cm² of filter area. For mycoplasma removal, 0.1 µm is used.
Does filtration kill microorganisms?
No. Filtration physically retains microorganisms on or in the filter; it does not kill them. The filter and any retained organisms must be inactivated separately (typically by autoclaving the used filter assembly).
Can filtration sterilize a virus-containing solution?
A 0.22 µm filter retains bacteria but not most viruses, which are typically 20–300 nm in diameter. Virus removal requires nanofiltration (typically 15–35 nm pore size) or other methods (heat, low-pH inactivation, gamma irradiation). Virus removal is a separate process step from bacterial sterilization.
How is a sterile filter validated?
Through three steps: bacterial retention validation against B. diminuta at ≥10⁷ CFU/cm², filter integrity testing (bubble point or diffusion test) on each use, and process validation covering fluid–filter compatibility and the full aseptic process per PDA TR26 and applicable GMP guidance.
When is autoclave sterilization preferred over filtration for liquids?
When the solution is heat-stable (water, simple buffers, salines, basic culture media), autoclaving is faster, cheaper, more thorough, and inactivates viruses. Filtration is the right choice when heat would degrade the product (proteins, vaccines, antibiotics, serum-containing media). See steam sterilization for autoclave guidance.
Can filter integrity testing miss a defective filter?
Bubble point and diffusion testing are validated non-destructive methods; if performed correctly with the specification matched to the filter and product, they reliably detect defects. Failure modes most often involve installation errors (gasket leaks, incorrect pre-wetting) rather than the test itself, which is why test procedures are part of the validated process.
Conclusion
Liquid filtration is the only practical sterilization method for heat-sensitive aqueous products and the standard for parenterals, vaccines, cell culture media, and a wide range of biologics. It removes bacteria but not most viruses or mycoplasma at 0.22 µm, and it depends on filter integrity rather than time–temperature kill — so validation under PDA TR26, USP, and GMP guidance is essential. For heat-stable liquids, steam sterilization is generally faster and more thorough; for non-liquid loads see the sterilization methods overview.