In recent years we have all seen the news articles and programs about problems with medical devices that were used on patients that were not adequately cleaned. Inadequate cleaning of medical devices is a complex issue that has many potential causes.
The Problem
Over the years as medical devices have become more complex the issues of how to perform acceptable cleaning, disinfection and sterilization have become greater. There are many stakeholders involved in cleaning, disinfection and sterilization of medical devices. Stakeholders include:
- regulatory agencies, FDA, CDC, state departments of public health;
- manufacturers of medical devices;
- reprocessors of medical devices;
- standards setting organizations; and
- test labs.
Who's Responsible?
The many stakeholders involved in this problem need to share the responsibility of ensuring that reprocessing procedures work and are followed. This process starts with the manufacturer of the medical device. When medical devices are developed the company has to submit the device for approval by the FDA before the device can be legally marketed in the USA. When developing a reusable device the manufacturer needs to prove that the device can be adequately cleaned and if needed disinfected or sterilized. After developing the reprocessing steps that need to be taken the manufacturer needs to validate those steps and then needs to write the IFUs (Instructions For Use) for the device user to follow. The processes for cleaning, disinfection and sterilization are frequently developed in conjunction with a test lab. Under laboratory conditions the test lab will verify and/or validate that the cleaning, disinfection or sterilization process is effective and achieves the goal of producing a cleaned, disinfected and/or sterilized device.
All of this information is submitted to the FDA as part of the device approval process. The FDA will review the reprocessing verification information, validation information and IFU and will either approve, disapprove or request more information. Once a device is approved the manufacturer is allowed to sell the device in the market place. The facility or person who purchases that device is then responsible for following the IFU.
The Causes
Reprocessing of medical devices sounds pretty straight forward, but unfortunately there are many things that can happen which end up with a device that has not been adequately cleaned and therefore cannot be disinfected or sterilized. In some cases it has been documented that the IFU for cleaning was not followed, causing a device to not be sterilized; and resulting in a healthcare associated infection (HAI). In other cases there are issues with following the IFU. Problems with cleaning medical devices include:
- the IFU is difficult to understand,
- the IFU is incomplete,
- the IFU is difficult (if not impossible) to follow,
- the person performing the cleaning process was not adequately trained, and
- the person performing the cleaning process did not follow the process.
Many people believe that if we follow the manufacturer’s instructions for use (IFU) on cleaning, disinfection and sterilization that there should be nothing to worry about.

Arthroscopic Shaver
Unfortunately there are many documented cases where the IFU was followed and the medical device was implicated in causing an infection. There was the case of a Texas hospital that had a series of infections caused by using a dirty instrument (an arthroscopic shaver). In this case one of the patients who got an infection had part of his shoulder bone and rotator cuff eaten away by the infection. Recently in 2014 and 2015 there have been several documented cases where duodenoscopes have been implicated in causing a HAI known as carbapenem-resistant Enterobacteriaceae (CRE). In all of these cases the manufacturer’s instructions were followed but the disinfection or sterilization process was not effective because following the instructions did not result in a clean instrument.

Duendoscope

Duendoscope tip is composed of small semi-enclosed spaces that are very difficult to clean
When it comes to cleanliness there is no current consensus on how to measure cleanliness or what level of cleanliness is acceptable. Sterility is typically defined as acceptable if the process results in a sterility assurance level (SAL) of 10-6. This SAL can only be reached if the device is adequately cleaned first. This point cannot be stressed enough. Recently there have been problems in which devices that were not adequately cleaned were disinfected and then these devices resulted in patients getting an HAI (see reference to duodenoscopes and CRE above). Choosing to sterilize these devices does not address the issue of inadequate cleaning. If a device is not clean then sterilizing it will not necessarily result in a sterile device. If during the cleaning process the microbial contamination on the device is not reduced to a level low enough for the sterilization process to work, then the device will not be sterilized. Both disinfection and sterilization rely on adequate cleaning. Sterilization does not override inadequate cleaning.
The format for creating an IFU is not standardized. This makes it difficult for reprocessing personnel to follow IFU’s. When the steps for reprocessing seem to jump around it is difficult for a person to keep straight what he/she is to do. Potentially steps are missed because of the lack of standardization. IFUs for virtually identical medical devices made by different companies can be very different which makes it difficult for reprocessing personnel to remember how to process a particular device.
Medical devices are sometimes developed to perform a particular function. During the manufacturing design phase emphasis is placed on two things: 1) how to make a device that will perform as desired and 2) what materials should the device be made from so that it will perform as desired. Unfortunately some devices have been designed without considering how the device will be reprocessed, disinfected and/or sterilized after it is used. This has resulted in device designs that are very difficult to clean, disinfect and/or sterilize. One only needs to look at the early designs for laparoscopic instrumentation, kerrison rongeurs and the current duodenoscope (to mention a few devices) to find a good example of this problem. Today laparoscopic instruments and kerrison rongeurs are being designed with cleaning and sterilization in mind and the new devices are much easier to clean. Unfortunately many of these early design laparoscopic instruments and rongeurs are still being used.

Single piece laparoscopic instrument

New style laparoscopic instrument that comes apart for cleaning

Single piece style kerrison rongeur
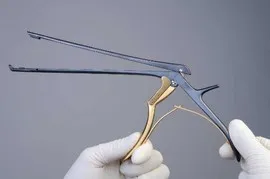
New style kerrison rongeur that opens for cleaning
Another aspect of a design problem involves developing a reprocessing procedure that is difficult to follow. Some IFU’s state that the medical device is supposed to be disassembled, then brushed with a small brush and at the end of the process reassembled. This can be difficult, if not impossible, when wearing protective attire. Decontamination gloves that provide adequate protection from sharps injury do not usually provide for being able to manipulate small components easily. Human factors need to be considered when designing a cleaning, disinfection or sterilization process.
A final area that needs to be looked at as a reprocessing challenge involves standards for training, education and certification. More complex processes for cleaning complex medical devices take more time to learn. For some devices demonstration of understanding the basics of a cleaning procedure is not adequate. Processing personnel need to be checked off and prove competence for cleaning of particular devices. Progress is being made in requiring certification of processing personnel. This is an area though where a lot more needs to be done. Unfortunately there are still many healthcare facilities that view sterile processing as an entry level position. In reality reprocessing of medical devices requires professionals who understand the concepts upon which practices are based and who are able to perform these complex tasks accurately, safely and skillfully.

Trying to pick up, inspect, disassemble, clean and reassemble medical devices and components can be difficult to do when wearing decontamination gloves
The Solutions
It is encouraging to see that progress on this issue is being made on several fronts. Professional organizations are working to develop standards that all stakeholders follow. In particular The Association for the Advancement of Medical Instrumentation (AAMI) has several working groups that are working to develop standards in several areas that address the challenges of reprocessing medical devices issue. These working groups include:
- Instructions for Reusable Device Reprocessing,
- Steam Sterilization Hospital Practices,
- Reusable Surgical Textiles Processing,
- Endoscope Reprocessing,
- Human Factors for Device Reprocessing,
- Quality Systems for Device Reprocessing, and
- Cleaning of Reusable Medical Devices.
Manufacturers now look at cleaning, disinfection and sterilization as part of device design. They are working with test labs to simulate “real life” situations when developing the cleaning and sterilization processes.The FDA is looking more closely at reprocessing issues when approving reusable medical devices. No longer are devices being approved that require extended sterilization exposure times. Manufacturers are being told that devices to be reprocessed must be designed that can be sterilized in standard sterilization cycles.
While these efforts are encouraging there is still the problem of devices that already exist and that continue to be sold. For these items device reprocessors need to be diligent to ensure that the devices are being adequately cleaned. This can be done by ensuring that manufacturer’s IFU’s are scrupulously followed. Emphasis needs to be placed on ensuring that employees perform return demonstrations to show that they understand how to perform a particular cleaning procedure. Devices need to be closely inspected after cleaning. There are many inspection devices available today, e.g. lighted magnifying glasses, magnifying cameras, telescopes for looking inside lumens, etc., which can be used to ensure devices are visibly clean. If a device is not visibly clean then it should not be disinfected or sterilized. If following the manufacturer’s IFU does not result in a visibly clean device, this should be reported to the healthcare facility’s infection control and risk management departments. Potentially this also needs to be reported to the FDA. Risk management usually makes that decision. In addition to inspecting devices for visible cleanliness the cleaning process should be subjected to cleaning verification. There are many products available today that can be used to verify that a device is clean and/or that a cleaning machine functions correctly. A comprehensive cleaning verification process needs to be part of the overall cleaning process for all devices.
Finally more needs to be done to provide quality education, training and certification for the people who are expected to reprocess medical devices. Reprocessing of medical devices is a difficult, complex job that needs to be recognized as such. More hospitals are now requiring the sterile processing staff to be certified but if we expect to have universal practices and adherence to complex procedures we need to have certification required by all healthcare facilities.
Conclusion
As can be seen there are many challenges to reprocessing medical devices. Improving these procedures requires that all involved parties work together so that devices can be developed that can be easily cleaned, disinfected and sterilized. This will take the cooperation of medical device manufacturers; test labs; regulatory agencies, e.g. FDA, CDC; professional organizations, e.g. AAMI, IAHCSMM, AORN; and reprocessing personnel.
Note: Pictures of the laparoscopic instruments and kerrison rongeurs used by permission of Carefusion V. Mueller. All other pictures are used by permission of Donna Swenson.
About author

Donna Swenson
Guest author
Donna Swenson, BS, CRCST, CHL, ACE, CSPDM, has more than 30 years of experience in the sterilization of medical devices, working with both healthcare providers and the medical device industry. For several years, Ms. Swenson has been the lead U.S. delegate to the ISO committee on moist heat sterilization of medical devices. In this position, she actively participated in the development of ANSI/AAMI/ISO 17665-1, -2, and -3; Sterilization of health care products – Moist heat – Part 1 Requirements, Part 2 Guidance and Part 3 Product Families. She is currently co-chair of the Industrial Moist Heat Sterilization of Medical Devices and Protective Barriers Committee. Ms. Swenson also serves on several other AAMI committees. She is also a member of AAMI’s job task analysis committee for the development of a certification program for Industrial Sterilization Scientists and is a member of the Editorial Board – Industrial Sterilization. Ms. Swenson is also a member of the Benchmarking Committee, co-sponsored by AAMI and IAHCSMM, and is a member of the Certification Council for IAHCSMM.
